 Synchrony Iterators (2020 - )
Synchrony Iterators (2020 - )
Modern programming languages provide comprehension syntax for manipulating collection types. Comprehension syntax makes programs more readable, but comprehensions typically correspond to nested loops. So, it is difficult using it to express efficient algorithms; I.e., a gap potentially exists in the intensional expressive power of comprehension syntax. The objectives of this project are to (i) prove that this intensional expressiveness gap is real; (ii) propose a programming construct, the Synchrony iterator, that precisely characterizes this gap; (iii) realise Synchrony iterator as a novel data type/class in some popular programming languages and dovetail it with comprehension syntax; (iv) show that Synchrony iterator generalizes merge-join in relational database management systems (RDBMS) to a large class of non-equijoin; and (v) integrate Synchrony iterator into an RDBMS (viz. PostgreSQL – a popular open-source RDBMS) so that its efficient merge-join can be generalized and used in executing more queries.
 Dealing with Confounders in Omics Analysis (2017 - )
Dealing with Confounders in Omics Analysis (2017 - )
Statistical feature selection on high-throughput omics data (e.g., genomics, proteomics, and transcriptomics) is commonly deployed to help understand the mechanism underpinning disease onset and progression. In clinical practice, these features are critical as biomarkers for diagnosis (see Glossary), guiding treatment, and prognosis. Unlike monogenic disorders, many challenging diseases (e.g., cancer) are polygenic, requiring multigenic signatures to counteract etiology and human variability issues. Unfortunately, in the course of analyzing omics data, we commonly encounter universality and reproducibility problems due to etiology and human variability, but also batch effects, poor experiment design, inappropriate sample size, and misapplied statistics. Current literature mostly blames poor experiment design and overreliance on the highly fluctuating P-value. In this project, we explore a deeper rethink on the mechanics of applying statistical tests (e.g. hypothesis statement construction, null distribution appropriateness, and test-statistic construction), and design analysis techniques that are robust on omics data.
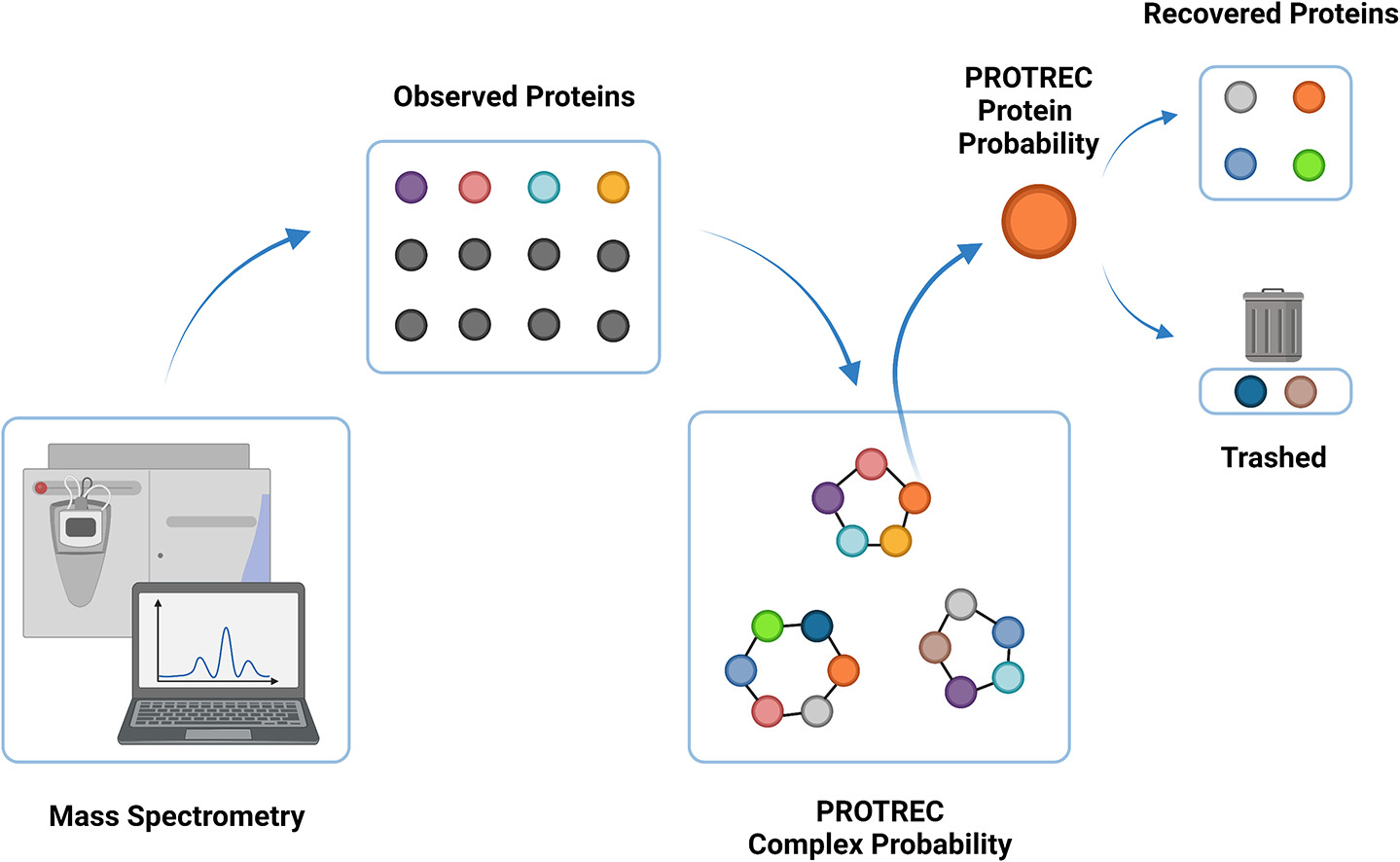 Recovering missing proteins based on Biological Complexes (2016 - )
Recovering missing proteins based on Biological Complexes (2016 - )
Advancements in proteomics are important to biological and clinical research because assaying protein identities/quantities paints an immediate picture of the underlying molecular landscape. However, proteomics still suffers from incomplete proteome coverage issues (i.e. not all proteins in a sample are observable in a single screen.) This gives rise to the "missing-protein problem" (MPP), which we define as difficulty in observing proteins in a proteome screen given that they are expected to be present. Due to MPP, efforts to extend proteome profiling in comparative or clinical studies, which require consistent protein/peptide detection (and accurate quantification across an extensive dynamic range), are rendered less effective. This project addresses the MPP based on a postulate that protein complexes provide a good context for making inference of a protein's presence and its abundance.
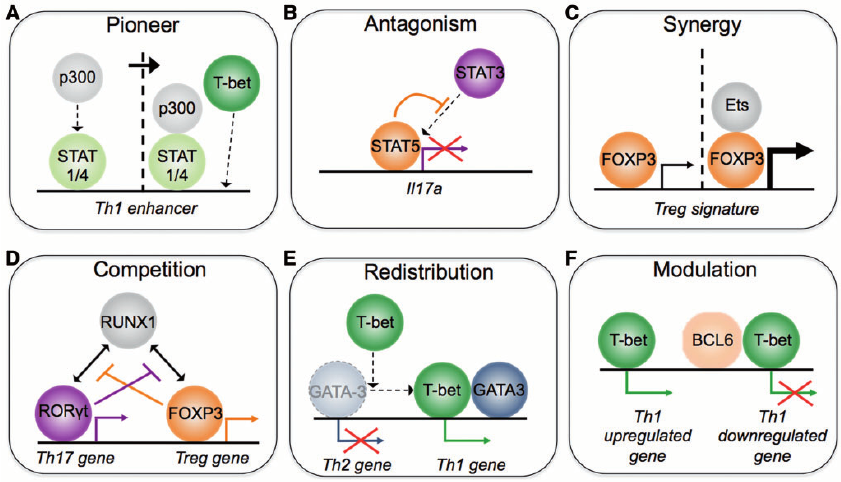 Transcription Factor-Interaction Prediction and Classification (2017 - )
Transcription Factor-Interaction Prediction and Classification (2017 - )
Transcriptional regulation is critical to the cellular processes of all organisms. Regulatory mechanisms often involve more than one transcription factor (TF) from different families, binding together and attaching to the DNA as a single complex, but only a fraction of the regulation partners of each transcription factor is currently known. It is thus of interest to biologists to analyse data extracted from TF-DNA binding experiments (e.g. ChIP-Seq) to infer the mechanisms that drive transcriptional regulation. In this project, we develop techniques for predicting the physical interaction between transcription factors, as well as for predicting the nature of their interactions (i.e. co-operative, competitive, or others).
 NGS Processing Technologies and Applications (2017 - )
NGS Processing Technologies and Applications (2017 - )
NGS (next-generation sequencing) technologies have become affordable for more applications. However, the data volumes produced also impose very high demands on computing resources to process and use. For example, genome assembly from NGS reads may take hours to complete, and may require processing nodes with terrabyte memory. This impedes applications in practical settings requiring fast turnaround and/or running on typical desktop computers. In this project, we develop technologies for more efficient processing of NGS data (e.g. compression, error correction, indexing, and assembly), and apply these technologies in real-life applications (e.g. neonatal precision medicine).
 Intensional Expressive Power of Query Languages (2012 - 2017)
Intensional Expressive Power of Query Languages (2012 - 2017)
Most existing studies on the expressive power of query languages have focused on what queries can be expressed and what queries cannot be expressed in a query language. They do not tell us much about whether a query can be implemented efficiently in a query language. Yet, paradoxically, efficiency is of primary concern in computer science. In contrast, the general goal of our proposed project is the development of powerful general methodology for studying the intensional expressive power of query languages, especially those that support nested relations, aggregate functions, powerset or recursion operations.
 Enabling More Sophisticated Proteomic Profile Analysis (2011 - 2017)
Enabling More Sophisticated Proteomic Profile Analysis (2011 - 2017)
Mass spectrometry (MS)-based proteomics is a powerful tool for profiling systems-wide protein expression changes. It can be applied for various purposes, e.g., biomarker discovery in diseases and study of drug responses. However, MS-based proteomics tend to have consistency (poor reproducibility and inter-sample agreement) and coverage (inability to detect the entire proteome) issues that need to be urgently addressed. In this project, we aim to deal with the two challenges above by proposing approaches that analyze proteomic profiles in the context of biological networks.
 Applications of Next-Generation Sequencing in Plants (2012 - 2015)
Applications of Next-Generation Sequencing in Plants (2012 - 2015)
Plant metabolites are compounds synthesized by plants for essential functions, such as growth and development (primary metabolites, such as lipid), and specific functions, such as pollinator attraction and defense against herbivores (secondary metabolites). Many of them are also used directly, or as derivatives, to treat a wide range of diseases for humans. There is thus much interest to study the biosynthesis of different plant metabolites and improve their yield. In this project, we apply next-generation sequencing techniques to to investigate lipid and secondary metabolisms, as well as to identify relevant DNA variations, in important crops.
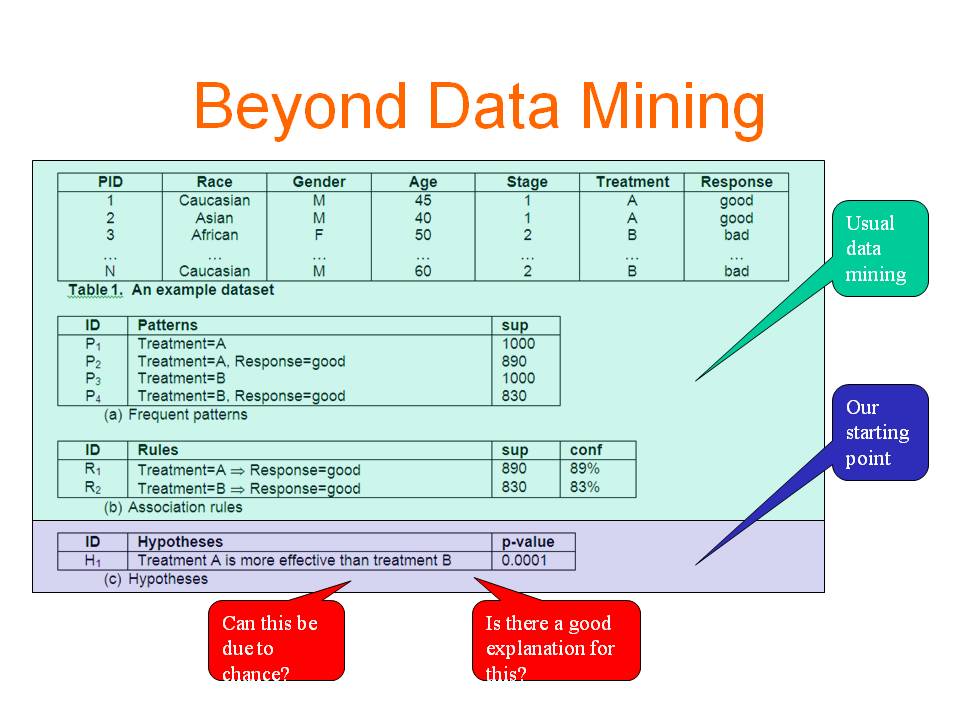 Exploratory Hypothesis Testing and Analysis (2010 - 2015)
Exploratory Hypothesis Testing and Analysis (2010 - 2015)
More and more data have been accumulated and stored in digital format in various applications. These data provide rich sources for making new discoveries. Data mining has become an important tool to transform data into knowledge. Finding useful and actionable knowledge is the main objective of diagnostic data mining. Most existing works tackle the problem by discovering patterns and rules and then studying their interestingness. In this work, we use a different paradigm which represents the discovered knowledge in the form of hypotheses. A hypothesis involves a comparison of two or more samples, which is more or less similar to how human obtain knowledge. Compared with patterns and rules, hypotheses provide the context in which a piece of information is interesting, thus hypotheses are more intuitive and informative than patterns and rules. More importantly, users can take actions more easily based on what a hypothesis indicates. We further analyse the discovered significant hypotheses and identify the reasons behind them so that users not only get to know what is happening but also have some rough ideas on when or why it is happening. This new data mining paradigm has the potential to make diagnostic data mining as successful as predictive data mining in real-life applications. In the proposed research, we will (1) formulate the problem and identify the issues that need to be addressed; (2) develop algorithms to solve the problem; (3) visualize the discovered knowledge to make the system easy to use; (4) interact and cooperate with domain experts in the biomedical area or other areas, and use the developed techniques to solve real-life problems.
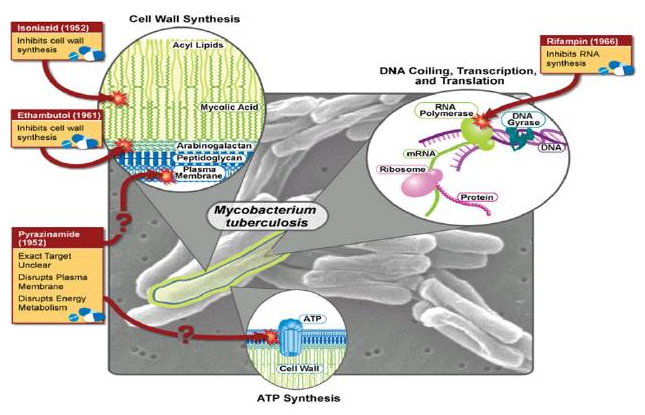 Reliable Protein Interactomes for Infectious Diseases (2010 - 2015)
Reliable Protein Interactomes for Infectious Diseases (2010 - 2015)
There is a critical need to address the emergence of drug resistant varieties of pathogens for several infectious diseases. For example, drug-resistant tuberculosis has continued to spread internationally and is now approaching critical proportions. Approaches to counter drug resistance have so far achieved limited success. It has been proposed that this lack of success is due to a lack of understanding of how resistance emerges in bacterial upon drug treatment and that a systems-level analysis of the proteins and interactions involved is essential to gaining insights into routes required for drug resistance. In this project, we propose to deal with the challenges in the systems-level analysis of proteins and interactions in pathogens of infectious diseases for identifying drug resistance pathways. Furthermore, we plan to use M. tuberculosis as a test case.
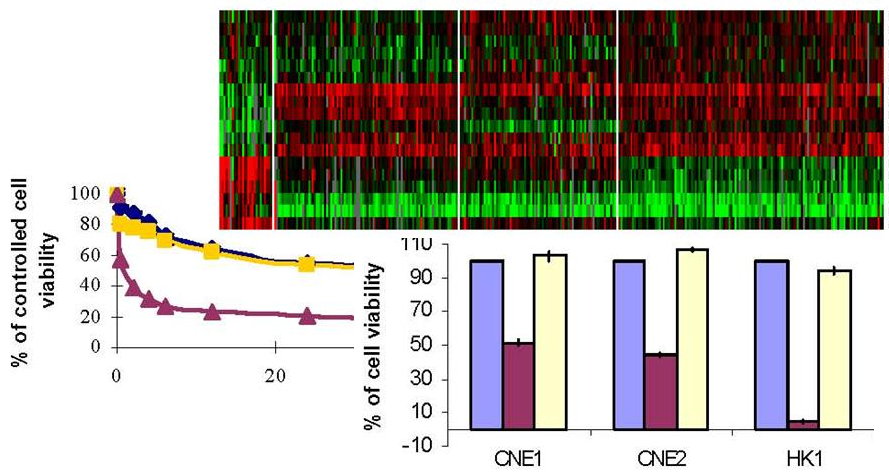 Enabling More Sophisticated Gene Expression Analysis (2007 - 2015)
Enabling More Sophisticated Gene Expression Analysis (2007 - 2015)
We find existing works on gene expression analysis fall short on several issues: these works provide little information on the interplay between selected genes; the collection of pathways that can be used, evaluated, and ranked against the observed expression data is limited; and a comprehensive set of rules for reasoning about relevant molecular events has not been compiled and formalized. We thus envision, in this project, a more advanced integrated framework to provide biologically inspired solutions for these challenges.
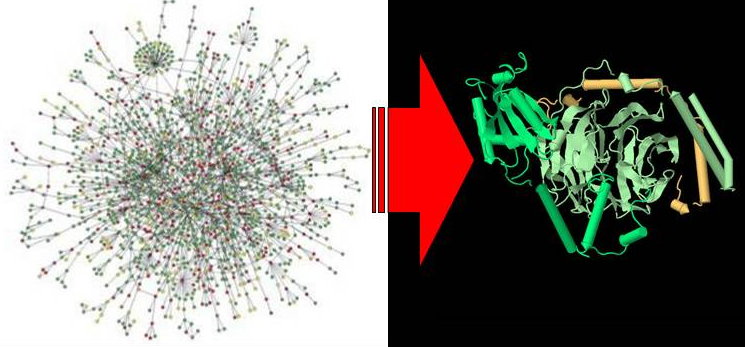 Protein Complex Prediction (2007 - 2015)
Protein Complex Prediction (2007 - 2015)
Protein interaction networks resulting from high-throughput assays are still essentially an in vitro scaffold. Further progress in computational analyses techniques and experimental methods is needed to reliably deduce in vivo protein interactions, to distinguish between permanent and transient interactions, to distinguish between direct protein binding from membership in the same protein complex and to distinguish protein complexes from functional modules. We hope to develop in this project a robust and powerful system to postprocess results of high-throughput PPI assays, as well as integrating extensive annotation information, to yield a more informative protein interactome beyond a mere in vitro scaffold.
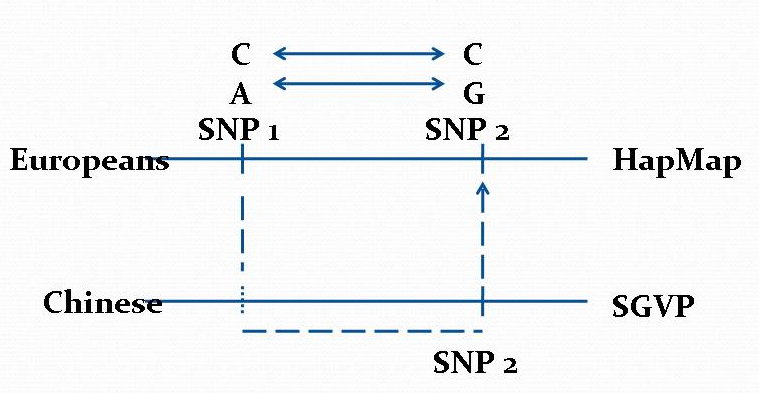 Data Mining Algorithms for Pharmacogenomics (2004 - 2013)
Data Mining Algorithms for Pharmacogenomics (2004 - 2013)
Human genome harbors millions of common single nucleotide polymorphisms (SNPs) and other types of genetic variations. These genetic variations play an important role in understanding the correlation between genetic variations and human diseases and the body's responses to prescribed drugs. The discovery of such genetic factors contributing to variations in drug response, efficiency, and toxicity has come to be known as pharmacogenomics. In this project, we explore several pharmacogenomic-related applications of database and datamining technologies.
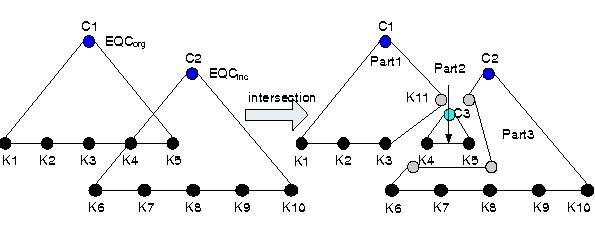 Pattern Spaces: Theory, Techniques, and Applications (2002 - 2010)
Pattern Spaces: Theory, Techniques, and Applications (2002 - 2010)
There are many previous data mining works on frequent itemsets, their closed patterns, and their generators. There are also a number of studies on emerging patterns and their borders. But the mining of odds ratio patterns, relative risk patterns, and patterns having other statistical properties frequently used in analysis of biomedical data, have never been investigated extensively. Thus odds ratio patterns, relative risk patterns, and patterns satisfying other statistical properties deserve our attention. In this project, we would like to (a) study in depth the theoretical properties of these patterns, (b) develop efficient algorithms for their mining, (c) develop efficient algorithms for their incremental maintenance when the underlying databases are updated, (d) investigate ways to build classifiers based on them and develop techniques for visualizing and explaining decisions made by such classifiers; and (e) to apply them to biomedical data.
 Individual-Based Modelling on the Spread of Infectious Diseases in Singapore
(2007 - 2009)
Individual-Based Modelling on the Spread of Infectious Diseases in Singapore
(2007 - 2009)
A large-scale epidemic simulation model of Singapore will be constructed in this project by taking demographic, social contact, and geographic factors into consideration. The project objectives include data collection, simulation and modelling, as well as strategy development for epidemic control.
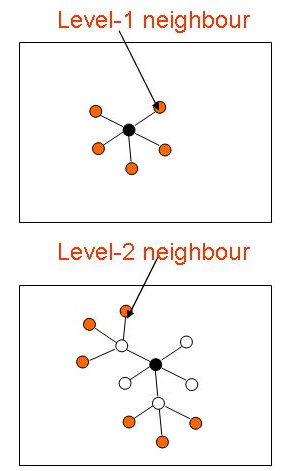 Graph-Based Protein Function Prediction (2005 - 2009)
Graph-Based Protein Function Prediction (2005 - 2009)
In this project, we investigate and develop graph-based methods for inferring protein functions without sequence homology. Most approaches in predicting protein function from protein-protein interaction data utilize the observation that a protein often share functions with proteins that interacts with it (its level-1 neighbors). However, proteins that interact with the same proteins (i.e. level-2 neighbors) may also have a greater likelihood of sharing similar physical or biochemical characteristics. We are interested to find out how significant is functional association between level-2 neighbors and how they can be exploited for protein function prediction. We will also investigate how to integrate protein interaction information with other types of information to improve the sensitivity and specificity of protein function prediction, especially in the absence of sequence homology.
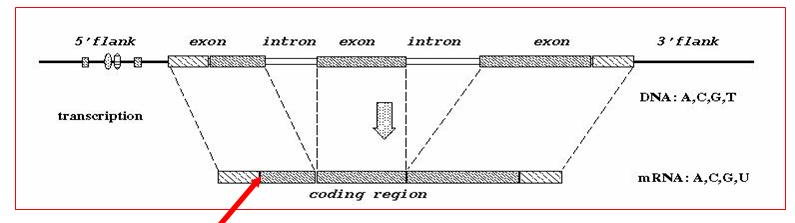 DNA Feature Recognition (2002 - 2008)
DNA Feature Recognition (2002 - 2008)
Correct prediction of transcription start sites, translation initiation sites, gene splice sites, poly-A sites, and other functional sites from DNA sequences are important issues in genomic research. In this project, we investigate these prediction problems using the paradigm of ``feature generation, feature selection, and feature integration''. There are two reasons for our interest in such a paradigm. The first reason is that standard tool boxes can be identified and used for each of the 3 components. For example, any statistical significance test can be used for feature selection. Similarly, any machine learning method can be used for feature integration. The main challenge is in developing a ``standard'' tool box for feature generation suitable for DNA functional sites. The second reason is that features that are critical to the recognition of specific DNA functional sites are explicitly generated and selected in this paradigm. This explicitness is helpful in understanding the underlying biological mechanism of that DNA functional site.
 GIORG NUS-ORI Data Collection System (2006 - 2007)
GIORG NUS-ORI Data Collection System (2006 - 2007)
The GIORG is a cross-hospitals study on gastro and intestinal cancer lead by NUS. There are 6 hospitals involved in the project. They are: National University Hospital (NUH), Singapore General Hospital (SGH), John Hopkins Hospital, Tan Tock Seng Hospital (TTSH), National Cancer Centre (NCC), and Alexandra Hospital. This project aims to manage all their patient information in a way that cross-hospital/-department web-based secure queries can be implemented easily. Previously, the data were stored in document files, and an NUS staff member has to go to the different hospitals to collect their data and key them into some MS excel files for further analysis. Many errors could be easily introduced in such a process. More importantly, because different hospitals were using different schemes to record patient information, it was almost impossible to retrieve "joint" data cross-hospitals. These issues lead us to develop the GIORG NUS-ORI Data Collection System here.
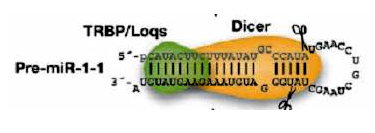 Recognition of MicroRNA Precursors and Targets (2005 - 2007)
Recognition of MicroRNA Precursors and Targets (2005 - 2007)
While the first miRNAs were discovered using experimental methods, experimental miRNA identification remains technically challenging and incomplete. This calls for the development of computational approaches to complement experimental approaches to miRNA gene identification. We propose in this project to investigate de novo miRNA precursor prediction methods. We follow the "generation, feature selection, and feature integration" paradigm of constructing recognition models for genomics sequences. We generate and identified features based on information in both primary sequence and secondary structure, and use these features to construct decision models for the recognition of miRNA precursors. In addition, analyzing the binding of miRNA to their mRNA target sites reveals that many different factors determine what constitutes a good fit. We thus intend to investigate these factors in detail and to construct decision models for predicting miRNA targets. Finally, we would like to understand the role of miRNAs in a number of human diseases. In particular, we plan to begin our analysis with genes involved in muscular dystrophy, as this group of genes are among the largest and most complex-structured human genes.
 Redundant Email Elimination (2000 - 2007)
Redundant Email Elimination (2000 - 2007)
With the advent of the Internet more and more people are communicating in electronic form such as via email messages, bulletin board systems and USENET groups. With this increased use of electronic communications, there has also become a greater degree of redundant messaging. Not only is it irritating for a person to find multiple repeated messages in their email folder, but it is a time consuming process for the person to read through all the messages and sort the relevant information from redundant messages. Thus we would like to develop an efficient and effective method and system to identify and eliminate such redundant messages.
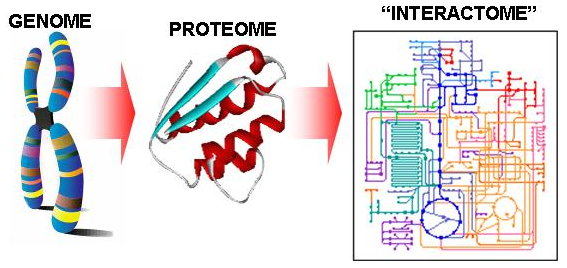 Increasing Confidence of Protein Interactomes (2003 - 2006)
Increasing Confidence of Protein Interactomes (2003 - 2006)
Progress in high-throughput experimental techniques in the past decade has resulted in a rapid accumulation of protein-protein interaction (PPI) data. However, recent surveys reveal that interaction data obtained by the popular high-throughput assays such as yeast-two-hybrid experiments may contain as much as 50% false positives and false negatives. As a result, further carefully-focused small-scale experiments are often needed to complement the large-scale methods to validate the detected interactions. However, the vast interactomes require much more scalable and inexpensive approaches. Thus it would be useful if the list of protein-protein interactions detected by such high-throughput assays could be prioritized in some way. Advances in computational techniques for assessing the reliability of protein-protein interactions detected by such high-throughput methods are explored in this project, especially those rely only on topological information of the protein interaction network derived from such high-throughput experiments.
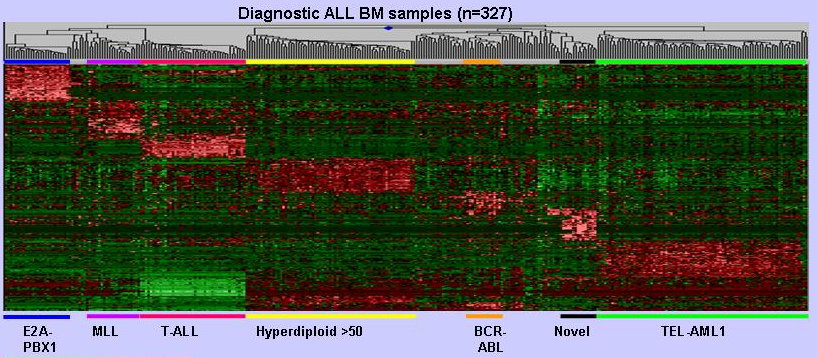 Tools for Design of Microarray and
Analysis of Gene Expression Profiles
for Disease Diagnosis and Prognosis (2000 - 2006)
Tools for Design of Microarray and
Analysis of Gene Expression Profiles
for Disease Diagnosis and Prognosis (2000 - 2006)
The development of microarray technology has made possible the simultaneous monitoring of the expression of thousands of genes. This development offers great opportunities in advancing the diagnosis of diseases, the treatment of diseases, and the understanding of gene functions. This project aims to: (1) develop technologies for the design of microarrays; (2) develop tools for the analysis of gene expression profiles, especially for optimization of disease treatment; and (3) apply these tools for optimization of disease treatment, with childhood leukemias as the initial area.
 The Protein Interaction Extraction System (1998 - 2002)
The Protein Interaction Extraction System (1998 - 2002)
A large part of the information required for biology research can only be found in free-text form, as in MEDLINE abstracts, or in comment fields of relevant reports, as in GenBank feature table annotations. This information is important for many types of analysis, such as classification of proteins into functional groups, discovery of new functional relationships, maintenance of information on material and methods, extraction of protein interaction information, and so on. However, information in free-text form is very difficult for automated systems to use. The project investigates techniques and applications of natural language processing to the extraction of biological information from free text.
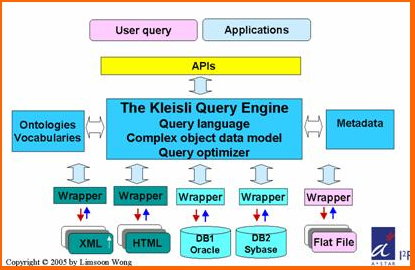 The Kleisli Query System (1994 - 2001)
The Kleisli Query System (1994 - 2001)
Kleisli is a data transformation and integration system that can be used for any application where the data is typed, and has proven especially useful for bioinformatics applications. It extends the conventional flat relational data model supported by the query language SQL to a complex object data model supported by the collection programming language CPL. It also opens up the closed nature of commercial relational data management systems to an easily extensible system that performs complex transformations on autonomous data sources that are heterogeneous and geographically dispersed.
 Querying Constraint Databases (1995 - 2000)
Querying Constraint Databases (1995 - 2000)
A relational database is traditionally formalized as a collection of finite relations. But this has limitation in some applications such as representing and querying spatial data. Thus, Kanellakis, Kuper, and Revesz in their famous PODS'90 paper introduce the idea of constraint databases and query languages for dealing with this type of data and queries. The original idea of constraint query languages is that quantifiers in a query can range over (say) the entire universe of real numbers, as opposed to only over those real numbers that occur in the underlying database. That is, constraint query languages permit the use of quantifiers under the ``natural domain semantics'' interpretation, while traditional database query languages interpret quantifiers strictly under the ``active domain semantics''. This ability to quantify over an infinite universe gives rise to two problems: (i) Queries now may have input/output that are infinite, so how would you represent them? (ii) How would you compute the queries if variables range over an infinite universe? These problems are solved by choosing an infinite universe with an underlying structure that allows an infinite set (on the universe) to be finitely representable and effectively computable. In this project, we study issues relating to constraint query languages where the underlying universe is the real closed field. The constraint query languages we study include extensions (with infinite sets and quantification over the real closed field) of relational calculus, SQL, and nested relational calculus. Towards the end of this project, we also venture into the ``embedded'' model theory of infinitary logics.
 Incremental SQL Queries (1995 - 1999)
Incremental SQL Queries (1995 - 1999)
Many queries such as transitive closure cannot be computed by SQL from scratch. However, many such queries can be maintained using SQL, given a previous answer and new record to be inserted or deleted. This phenomenon has an important practical implication, because the designer of a database application can plan for these type of queries by allocating some extra tables to stored their answers and maintaining these tables as records are inserted or deleted from the base tables. This project makes a thorough study of the expressive power of SQL and related query languages in such an ``incremental'' mode.
 Querying Nested Relations (1990 - 1997)
Querying Nested Relations (1990 - 1997)
We present a new principle for the development of database query languages that the primitive operations should be organized around types. Viewing a relational database as consisting of sets of records, this principle dictates that we should investigate separately operations for records and sets. There are two immediate advantages of this approach. Firstly, it provides a language for structures in which record and set types may be freely combined: nested relations or complex objects, Second, the fundamental operations for sets are closely related to those for other ``collection types'' such as bags and lists, and this suggests how database languages may be uniformly extended to these new types. We also look into various questions on the expressive power of query languages resulting from this paradigm. We prove a number of very fundamental results---the conservative extension property, the finite-cofiniteness property, and the bounded degree property---on nested relational query languages and on SQL.