


AI for Science
This research focuses on developing foundational artificial intelligence methods to advance scientific discovery. We study how modern machine learning models—such as deep learning, foundation models, and generative AI—can be tightly integrated with domain knowledge from physics, biology, and medicine to enable accurate modeling of biomolecular structures, interactions, and functions. This research direction is supported by national Foundational Research Capabilities (FRC) initiatives in Singapore, aiming to build long-term AI-driven scientific discovery platforms across disciplines.

- Bioinformatics Algorithms
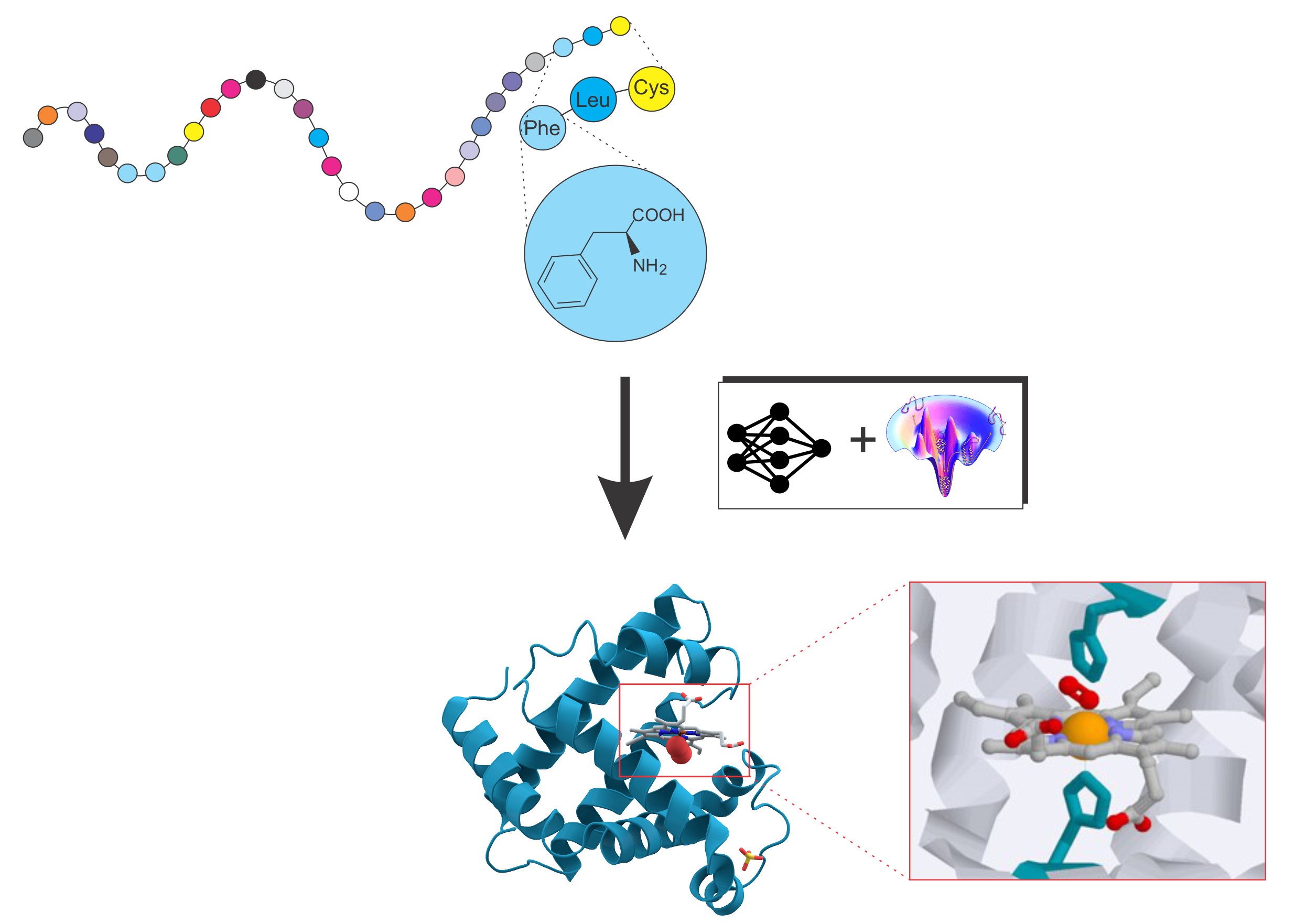
Protein folding and protein structure prediction
Deciphering the structure and function of proteins is central to modern biology and medicine. Our research develops pioneering computational frameworks that integrate advanced AI methodologies with physics-based force fields to accurately model protein structures and interactions. A central goal is to elucidate the fundamental relationships linking protein sequence, structure, dynamics, and function.
- Bioinformatics Algorithms
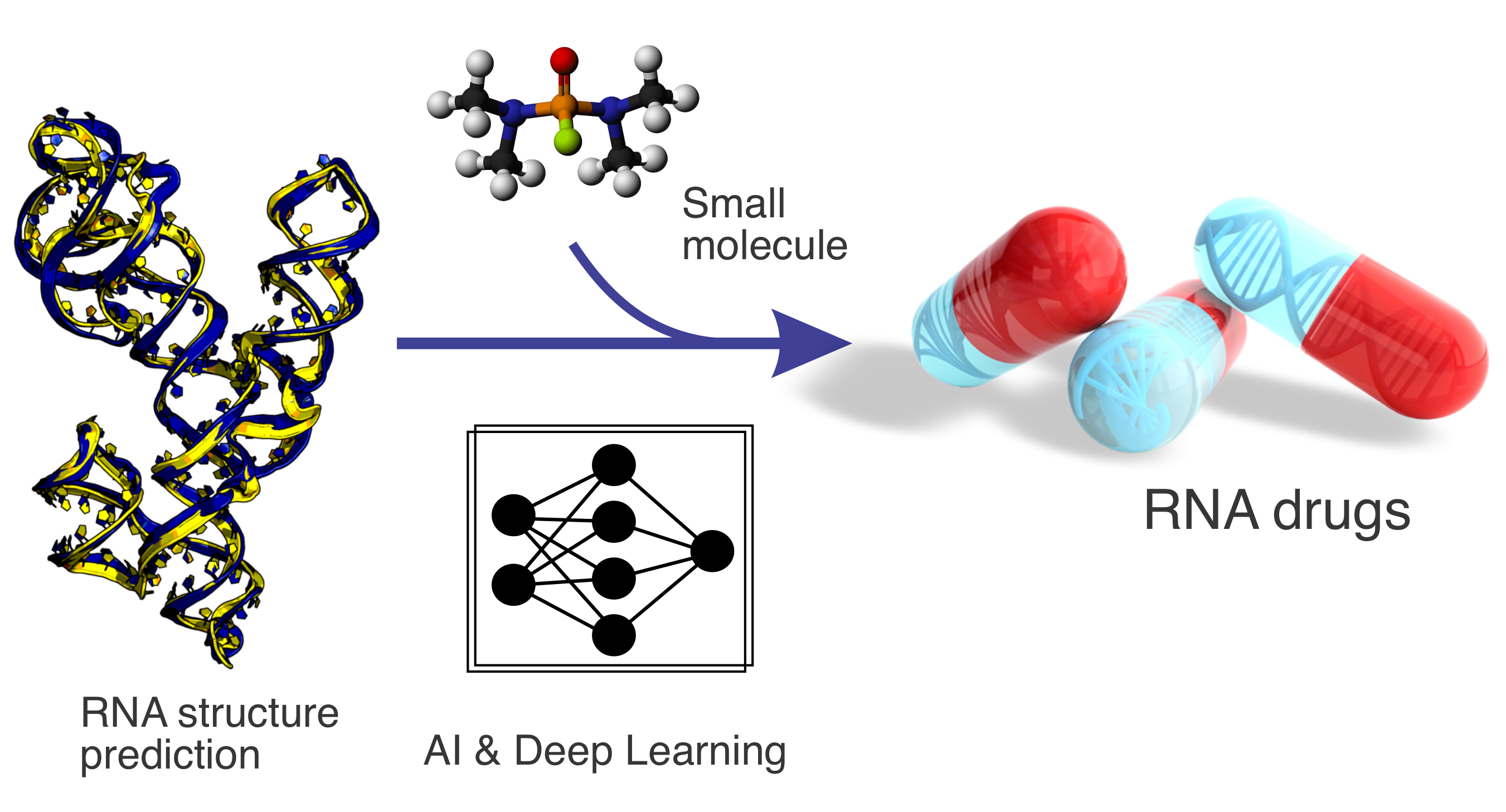
RNA structure prediction and small-molecule drug design
Non-coding RNAs play essential roles in regulating diverse cellular processes, yet their structural characterisation and ligand interactions remain challenging. This project develops advanced AI- and deep learning–based methods for RNA structure prediction and RNA–small-molecule interaction modeling. By integrating data-driven learning with biophysical principles, our work aims to enable RNA-targeted drug discovery and expand therapeutic opportunities beyond traditional protein-centric approaches.
- Bioinformatics Algorithms
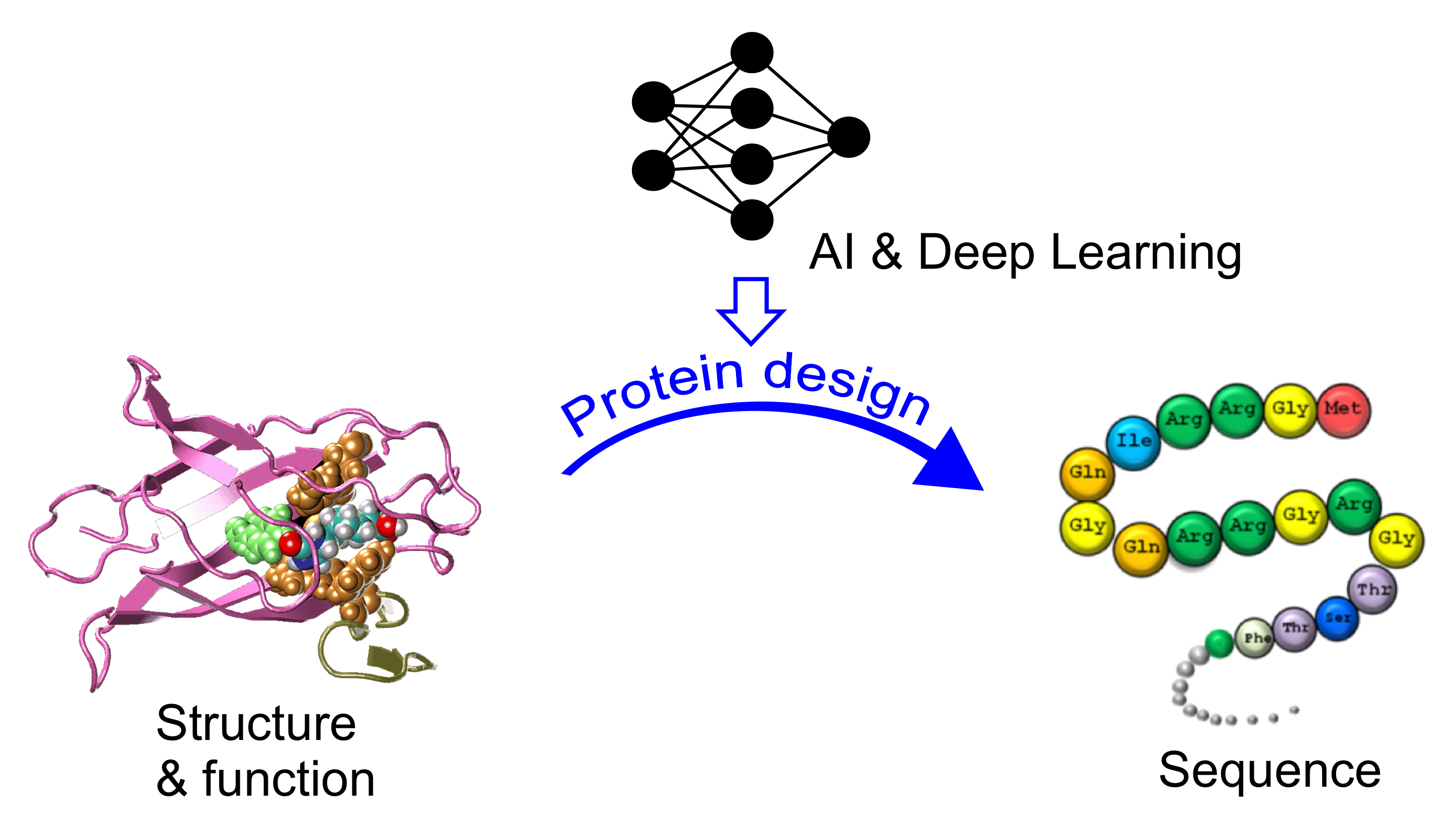
AI-based protein design and drug discovery
Natural proteins occupy a limited subset of the vast sequence–structure–function space shaped by evolution. This project develops AI- and deep learning–based frameworks for the rational design of novel proteins and peptides with tailored structures and functions. By integrating generative modeling with biophysical principles, we aim to expand the protein design space and enable new molecular scaffolds for biomedical and therapeutic applications.
- Bioinformatics Algorithms

From iteration on multiple collections in synchrony to fast general interval joins
Synchrony iterator captures a programming pattern for synchronized iterations. It is a conservative extension that enhances the repertoire of algorithms expressible in comprehension syntax. In particular, efficient general synchronized iterations, e.g. linear-time algorithms for low-selectivity database non-equijoins, become expressible naturally in comprehensinon syntax.
- TRL 4